
MEKTOVI 15 mg, comprimé pelliculé, boîte de 7 plaquettes de 12
Dernière révision : 14/11/2024
Taux de TVA : 2.1%
Prix de vente : 2 085,86 €
Taux remboursement SS : 100%
Base remboursement SS : 2 085,86 €
Laboratoire exploitant : PIERRE FABRE MEDICAMENT
Source :

Mélanome
Le binimetinib en association avec l'encorafenib est indiqué dans le traitement de patients adultes atteints de mélanome non résécable ou métastatique porteur d'une mutation BRAF V600
Cancer bronchopulmonaire non à petites cellules (CBNPC)
Le binimetinib en association avec l'encorafenib est indiqué dans le traitement des patients adultes atteints d'un cancer bronchopulmonaire non à petites cellules (CBNPC) avancé porteur d'une mutation BRAF V600E.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Le binimetinib doit être utilisé en association à l'encorafenib. Pour davantage d'informations sur les mises en garde et précautions associées au traitement par encorafenib, voir la rubrique Mises en garde spéciales et précautions d'emploi du RCP de l'encorafenib.
Le binimetinib en association à l'encorafenib chez les patients qui ont progressé sous inhibiteur de BRAF
Les données concernant l'association du binimetinib à l'encorafenib chez les patients ayant progressé sous un traitement antérieur par inhibiteur de BRAF pour un mélanome non résécable ou métastatique et porteurs d'une mutation BRAF V600 sont limitées. Ces données montrent que l'efficacité de cette association pourrait être réduite chez ces patients.
Binimetinib en association à l'encorafenib chez les patients présentant des métastases cérébrales
Les données d'efficacité chez les patients atteint d'un mélanome ou d'un CBNPC, porteur de la mutation BRAF V600 et présentant des métastases cérébrales traités par l'association de l'encorafenib au binimetinib sont limitées (voir rubrique Propriétés pharmacodynamiques).
Dysfonction ventriculaire gauche (DVG)
Une
dysfonction ventriculaire gauche, définie comme une diminution symptomatique ou
asymptomatique de la fraction d'éjection ventriculaire gauche (FEVG), peut
survenir lors du traitement par le binimetinib.
Il
est recommandé d'évaluer la FEVG par échocardiographie ou scintigraphie
myocardique (MUGA) avant l'initiation du traitement par binimetinib,
un mois après le début du traitement, puis à environ 3 mois d'intervalle ou
plus fréquemment au cours du traitement si cliniquement indiqué. La survenue
d'une diminution de la FEVG peut être prise en charge par une interruption du
traitement, une réduction de dose ou l'arrêt définitif du traitement (voir
rubrique Posologie et mode d'administration).
La tolérance du binimetinib en association à l'encorafenib n'a pas encore été établie chez les patients présentant une valeur initiale de FEVG inférieure à 50 %, ou inférieure à la limite inférieure de la normale selon les valeurs de références de l'hôpital. Par conséquent, chez ces patients, le binimetinib doit être utilisé avec prudence et en cas de DVG symptomatique, de FEVG de grade 3-4 ou de diminution d'au moins 10% de la FEVG en valeur absolue par rapport à la valeur initiale, le binimetinib doit être définitivement arrêté et la FEVG doit être ré-évaluée toutes les 2 semaines jusqu'à résolution.
Hémorragie
Des hémorragies, y compris des accidents hémorragiques majeurs, peuvent survenir lors de l'administration de binimetinib (voir rubrique Effets indésirables). Le risque hémorragique peut augmenter en cas d'utilisation concomitante d'un traitement anticoagulant et antiplaquettaire. La survenue d'accidents hémorragiques de grade ≥ 3 doit être prise en charge par la diminution de la dose, l'interruption temporaire du traitement ou l'arrêt définitif (voir Tableau 2 à la rubrique Posologie et mode d'administration) et selon le tableau clinique.
Toxicités oculaires
Des toxicités oculaires, notamment décollement de l'épithélium pigmentaire de la rétine (DEPR) et occlusion de la veine rétinienne (OVR), peuvent survenir lors de l'administration de binimetinib. Des cas d'uvéite (dont iridocyclite et iritis) ont été rapportés chez des patients traités par binimetinib en association avec l'encorafenib (voir rubrique Effets indésirables).
Le binimetinib n'est pas recommandé chez les patients ayant des antécédents d'OVR. La tolérance du binimetinib n'a pas été établie chez les patients présentant des facteurs prédisposants à l'OVR, notamment un glaucome non contrôlé, une hypertension oculaire, un diabète incontrôlé ou des antécédents de syndrome d'hyperviscosité ou d'hypercoagulabilité. Par conséquent, le binimetinib doit être utilisé avec précaution chez ces patients.
À
chaque visite, les symptômes visuels doivent être évalués chez les patients. Si
des symptômes indiquant l'apparition ou l'aggravation de troubles visuels
notamment une baisse de la vision centrale, une vision trouble ou une perte de
la vue sont observés, il est recommandé de procéder rapidement à un examen
ophtalmologique.
La
survenue d'un décollement de l'épithélium pigmentaire de la rétine peut être
prise en charge par une interruption du traitement, une réduction de la dose ou
l'arrêt définitif du traitement (voir Tableau 1 à la rubrique Posologie et
mode d'administration).
Le
binimetinib doit être définitivement arrêté en cas de
survenue d'une Occlusion de la Veine Rétinienne. (Voir Tableau 1 à la rubrique Posologie
et mode d'administration).
En cas de survenue d'une uvéite pendant le traitement, se référer à la rubrique Posologie et mode d'administration du RCP de l'encorafenib pour plus d'informations.
Élévation des taux de CPK et rhabdomyolyse
Une élévation des taux de CPK a été rapportée chez les patients traités par le binimetinib (voir rubrique Effets indésirables) ; des cas de rhabdomyolyse ont été rapportés peu fréquemment. Une attention particulière devra être portée aux patients atteints de troubles neuromusculaires associés à l'élévation des CPK et à la rhabdomyolyse.
Les taux de CPK et de la créatinine doivent être contrôlés tous les mois pendant les 6 premiers mois de traitement et en fonction du tableau clinique. Les patients doivent être avisés de maintenir une bonne hydratation pendant le traitement. Selon la sévérité des symptômes, le degré d'élévation des taux de CPK ou de la créatinine, une réduction de dose, une interruption de dose ou l'arrêt définitif du binimetinib peuvent être nécessaires (voir le Tableau 1 à la rubrique Posologie et mode d'administration).
Hypertension
Une hypertension, ou l'aggravation d'une hypertension préexistante, peut se produire lors de l'utilisation de binimetinib. La pression artérielle doit être mesurée à l'initiation du traitement et au cours du traitement, et toute hypertension doit être contrôlée par les thérapies usuelles, le cas échéant.
En cas d'hypertension artérielle sévère, une interruption temporaire du binimetinib est recommandée jusqu'à ce que l'hypertension soit contrôlée (voir le Tableau 2 de la rubrique Posologie et mode d'administration).
Maladie thromboembolique (MTEV)
Une
maladie thromboembolique veineuse peut survenir lors d'un traitement par binimetinib (voir rubrique Effets indésirables). Le binimetinib doit être utilisé avec prudence chez les
patients à risque de maladie thromboembolique veineuse ou ayant des antécédents
thromboemboliques veineux.
En
cas de thrombose veineuse profonde ou d'embolie pulmonaire survenue pendant le
traitement, il conviendra de recourir à une réduction de dose, une interruption
de dose ou à un arrêt définitif du traitement (voir Tableau 1 à la rubrique Posologie
et mode d'administration).
Pneumopathie inflammatoire/Pneumopathie interstitielle diffuse
Une pneumopathie inflammatoire/pneumopathie interstitielle diffuse peut apparaître lors du traitement par binimetinib. Le traitement par binimetinib doit être interrompu chez les patients présentant une pneumopathie inflammatoire ou une pneumopathie interstitielle diffuse suspectée, notamment chez les patients présentant des symptômes pulmonaires nouveaux ou évolutifs, comme toux, dyspnée, hypoxie, opacités réticulées ou infiltrats parenchymateux (voir Tableau 1 à la rubrique Posologie et mode d'administration). Le binimetinib doit être définitivement arrêté chez les patients chez lesquels un diagnostic de pneumopathie interstitielle diffuse a été posé.
Nouvelles tumeurs primitives
De nouvelles tumeurs primitives, cutanées et non cutanées, ont été observées chez des patients traités par inhibiteurs de BRAF et peuvent apparaître lors de l'administration du binimetinib en association à l'encorafenib (voir rubrique Effets indésirables).
Tumeurs cutanées
Des
tumeurs cutanées, telles qu'un carcinome épidermoïde cutané (CEC), incluant des
cas de kératoacanthome, ont été observées chez des
patients traités par binimetinib lorsqu'il est
utilisé en association à l'encorafenib.
Un
examen clinique dermatologique doit être effectué avant le début du traitement
par binimetinib en association à encorafenib,
tous les 2 mois pendant le traitement et jusqu'à 6 mois maximum après l'arrêt
de celui-ci. Les lésions suspectes de la peau doivent être traitées par exérèse
cutanée avec un examen dermato-anatomopathologique. Les patients doivent être
avertis que dans ces cas, ils doivent immédiatement signaler à leur médecin
l'apparition de toute nouvelle lésion cutanée. Le traitement par le binimetinib en association à l'encorafenib
peut être poursuivi sans modifications de dose.
Tumeurs non cutanées
En
raison de son mécanisme d'action, l'encorafenib peut
favoriser l'apparition de tumeurs associées à l'activation du gène RAS via des
mutations ou d'autres mécanismes. Les patients recevant le binimetinib
en association à l'encorafenib doivent bénéficier
d'un examen clinique de la tête et du cou, d'un scanner thoraco-abdominal et
d'un examen de la région anale et pelvienne (pour les femmes), ainsi que d'une
numération de formule sanguine complète avant le début, au cours et à la fin du
traitement, comme cliniquement approprié.
L'arrêt
définitif du binimetinib et de l'encorafenib
doit être envisagé chez les patients qui développent des tumeurs non cutanées
avec mutation du gène RAS. Les avantages et les risques doivent être évalués
attentivement avant d'administrer le binimetinib en
association à l'encorafenib aux patients ayant des
antécédents de cancer ou atteints actuellement d'un cancer associé à une
mutation du gène RAS.
Syndrome de lyse tumorale (SLT)
La survenue de SLT, qui peut être fatale, a été associée à l'utilisation du binimetinib en association avec l'encorafenib (voir rubrique Effets indésirables). Les facteurs de risque du SLT comprennent une charge tumorale élevée, une insuffisance rénale chronique préexistante, une oligurie, une déshydratation, une hypotension et une urine acide. Ces patients doivent être étroitement surveillés et traités rapidement selon les indications cliniques, et une hydratation prophylactique doit être envisagée.
Anomalies biologiques hépatiques
Des anomalies biologiques hépatiques, notamment une élévation des transaminases ASAT et ALAT peuvent survenir avec le binimetinib (voir rubrique Effets indésirables). Les paramètres biologiques hépatiques doivent faire l'objet d'un contrôle avant le début du traitement par binimetinib et encorafenib et faire l'objet d'une surveillance au moins mensuelle pendant les 6 premiers mois de traitement, puis si cliniquement indiqué. Les anomalies biologiques hépatiques doivent être prises en charge avec une réduction de dose, une suspension de dose ou l'arrêt définitif du traitement (voir Tableau 1 de la rubrique Posologie et mode d'administration).
Insuffisance hépatique
Le métabolisme hépatique principalement via glycurono-conjugaison est la principale voie d'élimination du binimetinib (voir rubrique Propriétés pharmacocinétiques). L'encorafenib étant déconseillé chez les patients présentant une insuffisance hépatique modérée (Child-Pugh B) et sévère (Child-Pugh C), l'administration du binimetinib n'est pas recommandée dans ces cas (voir les rubriques Posologie et mode d'administration et Propriétés pharmacocinétiques).
Intolérance au lactose
Mektovi contient du lactose. Les rares patients souffrant d'intolérance héréditaire au lactose, déficit total en lactase ou malabsorption du glucose/galactose ne doivent pas prendre ce médicament.
Résumé du profil de sécurité
La
sécurité du binimetinib (45 mg par voie orale deux fois par jour) en
association avec l'encorafenib (450 mg par voie orale une fois par jour) a été évaluée
dans la population de sécurité intégrée (ISP) de 372 patients, incluant des
patients atteints de mélanome non résécable ou métastatique porteurs d'une
mutation BRAF V600E et des patients atteints d'un CBNPC avancé porteurs d'une
mutation BRAF V600E (ci-après dénommé ISP Combo450). Dans l'ISP Combo 450, 274
patients ont reçu l'association pour le traitement du mélanome non résécable ou
métastatique porteurs d'une mutation BRAF V600 (dans deux études de phase II
(CMEK162X2110 et CLGX818X2109) et une étude de phase III (CMEK162B2301, partie
1) et 98 patients ont reçu l'association pour le traitement du CBNPC avancé
porteur d'une mutation BRAF V600E (dans une étude de phase II non randomisée
(ARRAY-818-202)) (voir rubrique Propriétés pharmacodynamiques).
Les
effets indésirables les plus fréquents (≥ 25 %) observés chez les
patients traités par du binimetinib en association à l'encorafenib ont été :
fatigue, nausées, diarrhée, vomissements, douleurs abdominales, myopathies /
troubles musculaires et arthralgies.
La sécurité de l'encorafenib (300 mg par voie orale une fois par jour) associé au binimetinib (45 mg par voie orale deux fois par jour) a été évaluée chez 257 patients atteints de mélanome métastatique ou non résécable à gène BRAF présentant la mutation V600 (ci-après désigné comme la population COMBO 300), selon l'étude de phase III (CMEK162B2301, partie 2). Les effets indésirables les plus fréquents (> 25 %) survenus chez les patients traités par encorafenib 300 mg en association au binimetinib étaient les suivants : fatigue, nausées et diarrhée.
Liste des effets indésirables sous forme de tableau
Les effets indésirables sont énumérés ci-dessous selon la classe de système d'organes MedDRA et la convention de fréquence suivante : Très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Pour chaque catégorie de fréquence, les effets indésirables sont présentés en ordre décroissant de gravité.
Tableau 3 : Effets indésirables survenant chez des patients recevant le binimetinib en association à l'encorafenib à la dose recommandée (n = 372)| Classe de système d'organes | Effets indésirables |
Fréquence
(tous grades confondus) |
|
Tumeurs
bénignes, malignes et non précisée |
Carcinome épidermoïde cutanéa | Fréquent |
| Papillome cutané* | Fréquent | |
| Carcinome basocellulaire* | Peu fréquent | |
|
Affections
hématologiques et du système lymphatique |
Anémie | Très fréquent |
|
Affections
du système immunitaire |
Hypersensibilitéb | Fréquent |
|
Troubles
du métabolisme et de la nutrition |
Syndrome de lyse tumorale | Indéterminée |
|
Affections
du système nerveux |
Neuropathie périphérique* | Très fréquent |
| Sensations vertigineuses* | Très fréquent | |
| Céphalées* | Très fréquent | |
| Dysgueusie | Fréquent | |
| Parésie facialec | Peu fréquent | |
| Affections oculaires | Troubles de la vision* | Très fréquent |
|
Décollement
de l'épithélium pigmentaire de la rétine (DEPR)* |
Très fréquent | |
| Uvéite* | Fréquent | |
| Affections cardiaques |
Dysfonction
ventriculaire gauche (DVG)d |
Fréquent |
| Affections vasculaires | Hémorragiee | Très fréquent |
| Hypertension* | Très fréquent | |
|
Maladie
thromboembolique veineuse (MTEV)f |
Fréquent | |
|
Affections
gastro- intestinales |
Douleur abdominale* | Très fréquent |
| Diarrhée* | Très fréquent | |
| Vomissements* | Très fréquent | |
| Nausées | Très fréquent | |
| Constipation | Très fréquent | |
| Coliteg | Fréquent | |
| Pancréatite* | Peu fréquent | |
|
Affections
de la peau et du tissu sous-cutané |
Hyperkératose* | Très fréquent |
| Rash* | Très fréquent | |
| Sécheresse cutanée* | Très fréquent | |
| Prurit* | Très fréquent | |
| Alopécie* | Très fréquent | |
| Photosensibilité* | Fréquent | |
| Dermatite acnéiforme* | Fréquent | |
| Érythrodysesthésie palmo-plantaire (EPP) | Fréquent | |
| Érythème* | Fréquent | |
| Panniculite* | Fréquent | |
|
Affections musculo-squelettiques et systémiques |
Arthralgies* | Très fréquent |
| Myopathie/Troubles musculairesh | Très fréquent | |
| Dorsalgie | Très fréquent | |
| Douleurs aux extrémités | Très fréquent | |
| Rhabdomyolyse | Peu fréquent | |
|
Affections
du rein et des voies urinaires |
Insuffisance rénale* | Fréquent |
|
Troubles
généraux et anomalies au site d'administration |
Fièvre* | Très fréquent |
| Œdème périphériquei | Très fréquent | |
| Fatigue* | Très fréquent | |
| Investigations |
Créatine
phosphokinase plasmatique augmentée |
Très fréquent |
| Transaminases augmentées* | Très fréquent | |
| Gamma-glutamyltransférase (GGT) augmentée* | Très fréquent | |
| Créatinine sanguine augmentée* | Fréquent | |
| Phosphatase alcaline sanguine augmentée | Fréquent | |
| Amylase augmentée | Fréquent | |
| Lipase augmentée | Fréquent |
* noms composés qui
comprenaient plusieurs termes préférés
a comprend kératoacanthome, carcinome épidermoïde et carcinome épidermoïde de la peau
b comprend notamment, œdème de Quincke, hypersensibilité médicamenteuse, hypersensibilité, vascularite d'hypersensibilité et urticaire
c comprend trouble lié au nerf facial, paralysie faciale, parésie faciale, paralysie de Bell
d comprend dysfonction ventriculaire gauche, fraction d'éjection ventriculaire gauche diminuée, insuffisance cardiaque et fraction d'éjection ventriculaire anormale
e comprend les hémorragies à différents sites, notamment les hémorragies cérébrales, hémorragies intracrâniennes, hémorragies vaginales, saignements menstruels abondants, saignements intermenstruels, hématochézie, hémoptysie, hémothorax, hémorragies gastro-intestinales et hématurie.
f comprend notamment, embolie pulmonaire, thrombose veineuse profonde, embolie, thrombophlébite, thrombophlébite superficielle, thrombose, phlébite, syndrome de la veine cave supérieure, thrombose de la veine mésentérique et thrombose de la veine cave.
g comprend colite, colite ulcéreuse, entérocolite et proctite
h comprend myalgies, faiblesse musculaire, spasme musculaire, lésion musculaire, myopathie, myosite i comprend notamment, rétention hydrique, œdème périphérique, œdème localisé, oedème généralisé et enflure.
a comprend kératoacanthome, carcinome épidermoïde et carcinome épidermoïde de la peau
b comprend notamment, œdème de Quincke, hypersensibilité médicamenteuse, hypersensibilité, vascularite d'hypersensibilité et urticaire
c comprend trouble lié au nerf facial, paralysie faciale, parésie faciale, paralysie de Bell
d comprend dysfonction ventriculaire gauche, fraction d'éjection ventriculaire gauche diminuée, insuffisance cardiaque et fraction d'éjection ventriculaire anormale
e comprend les hémorragies à différents sites, notamment les hémorragies cérébrales, hémorragies intracrâniennes, hémorragies vaginales, saignements menstruels abondants, saignements intermenstruels, hématochézie, hémoptysie, hémothorax, hémorragies gastro-intestinales et hématurie.
f comprend notamment, embolie pulmonaire, thrombose veineuse profonde, embolie, thrombophlébite, thrombophlébite superficielle, thrombose, phlébite, syndrome de la veine cave supérieure, thrombose de la veine mésentérique et thrombose de la veine cave.
g comprend colite, colite ulcéreuse, entérocolite et proctite
h comprend myalgies, faiblesse musculaire, spasme musculaire, lésion musculaire, myopathie, myosite i comprend notamment, rétention hydrique, œdème périphérique, œdème localisé, oedème généralisé et enflure.
Lorsque l'encorafenib était administré à une dose de 300 mg une fois par jour en association au binimetinib à 45 mg deux fois par jour (COMBO300) dans l'étude CMEK162B2301, partie 2, la catégorie de fréquence était inférieure par rapport à l'ensemble de la population COMBO450 pour les effets indésirables suivants : anémie, neuropathie périphérique, hémorragie, hypertension, prurit (fréquent) et colite, ainsi qu'augmentation de l'amylase et de la lipase (peu fréquent).
Description de certains effets indésirables
Tumeurs cutanées
Un carcinome épidermoïde cutané a été rapporté lorsque le binimetinib était
utilisé en association à l'encorafenib (voir la rubrique Effets indésirables
du RCP de l'encorafenib).
Atteintes oculaires
Dans l'ISP Combo450, un décollement de l'épithélium pigmentaire de la rétine a
été signalé chez 22,3 % des patients (83/372). Le décollement de l'épithélium pigmentaire de la
rétine était de grade 1 (asymptomatique) chez 15,6 % des patients (58/372), de
grade 2 chez 5,1 % des patients (19/372) et de grade 3 chez 1,6 % des patients
(6/372).
La plupart des événements ont été rapportés comme rétinopathie, décollement de
la rétine, liquide sous-rétinien, œdème maculaire et chorio-rétinopathie
séreuse centrale et ont entraîné des interruptions de dose ou des modifications
de dose chez 3,8 % des patients (14/372). Le délai médian d'apparition du
premier événement de décollement de l'épithélium pigmentaire de la rétine (tous
grades confondus) était de 1,4 mois (allant de 0,00 à 17,5 mois).
Des troubles visuels, y compris vision trouble et baisse de l'acuité visuelle,
sont survenus chez 23,1 % des patients (86/372). Les troubles visuels ont
généralement été réversibles.
L'uvéite a été également rapportée lorsque le binimetinib était utilisé en association à l'encorafenib (voir la rubrique Effets indésirables du RCP de l'encorafenib).
Dans l'étude CMEK162B2301, partie 2, bras COMBO300, un décollement de l'épithélium pigmentaire de la rétine est survenu chez 12,5 % des patients (32/257), un grade 4 étant observé chez 0,4 % des patients (1/257).
Dysfonction ventriculaire gauche
Dans l'ISP COMBO450, une dysfonction ventriculaire gauche a été rapportée chez
9,4 % des patients (35/372). Des événements de grade 3 sont survenus chez 1,3 %
des patients (45/372). La dysfonction ventriculaire gauche a nécessité des
interruptions du traitement chez 0,84 % des patients (3/372) et des réductions
de dose chez 6,2 % des patients (23/372).
Le délai médian jusqu'à la première survenue d'une dysfonction ventriculaire
gauche (tous grades confondus) était de 5,2 mois (allant de 0,0 à 25,7 mois)
chez les patients ayant développé une FEVG inférieure à 50 %. La valeur moyenne
des FEVG a diminué de 5,3 % dans l'ISP Combo450, passant d'une moyenne de 63,3
% à l'inclusion à 58,0 %. La dysfonction ventriculaire gauche a généralement
été réversible après réduction de dose ou interruption du traitement.
Hémorragie
Des événements hémorragiques ont été observés chez 16,7 % des patients (62/372)
dans l'ISP Combo450. La plupart de ces événements étaient de grade 1 ou 2 :
13,2 % (49/372) et 3,5 % (13/372) étaient de grade ≥ 3. Peu de patients
ont nécessité une interruption du traitement ou une réduction de dose (2,4 %,
soit 9/372). Les événements hémorragiques ont conduit à l'arrêt définitif du
traitement chez 0,8 % des patients (3/372). Les événements hémorragiques les
plus fréquents étaient hématurie chez 2,7 % des patients (10/372), hématochézie
chez 2,7 % des patients (10/372), et rectorragie chez 2,2 % des patients
(8/372) . Une hémorragie fatale due à un ulcère gastrique avec une défaillance
multiviscérale comme cause simultanée de décès, a été rapportée chez un
patient. Une hémorragie cérébrale : hémorragie intracrânienne est survenue chez
1,3 % des patients (5/372), d'évolution fatale chez 4 patients. Tous les
événements sont survenus dans le contexte de nouvelles métastases cérébrales ou
évolutives.
Dans l'étude CMEK162B2301, partie 2, bras COMBO300, des événements hémorragiques sont survenus chez 6,6 % des patients (17/257), avec un grade 3-4 observé chez 1,6 % des patients (4/257).
Hypertension
L'apparition d'une hypertension ou l'aggravation d'une hypertension
préexistante ont été rapportées chez 11,0 % (41/372) des patients traités avec
ISP Combo450. L'hypertension rapportée était de
grade 3 chez 5,1 % des patients (19/372), y compris crise hypertensive (0,3 %
(1/372)). L'hypertension a conduit à une interruption ou une modification de
dose chez 2,2 % (8/372) des patients et a nécessité un traitement
complémentaire chez 7,5 % des patients (28/372).
Maladie thromboembolique veineuse (MTEV)
Chez l'ISP Combo450, une MTEV est survenue chez 4,8 % des patients (18/372),
dont 1,9 % des patients (7/372) ayant présenté une embolie pulmonaire. La METV
était de grade 1 ou 2 chez 4,0 % des patients (15/372) et de grade 3 ou 4 chez
0,8 % des patients (3/372). La METV a conduit à une interruption du traitement ou
une modification de dose chez 1,1 % des patients (4/372) et à un traitement
complémentaire chez 4,6 % des patients (17/372).
Pancréatite
Des cas de pancréatite ont été rapportés lors du traitement par le binimetinib
en association à l'encorafenib (voir la rubrique Effets indésirables du
RCP de l'encorafenib).
Réactions cutanées
Des réactions cutanées peuvent survenir lorsque le binimetinib est utilisé en
association à l'encorafenib.
Rash
Dans l'ISPOMBO Combo450, un rash est survenu chez 20,4 % des patients (76/372).
La plupart des événements étaient sans sévérité, avec des événements de grade 3
ou 4 rapportés chez 1,1 % des patients (4/372). Le rash a conduit à l'arrêt du
traitement chez 0,8 % des patients (3/372) et à une interruption ou une
modification de la dose chez 2,4 % des patients (9/372).
Dermatite acnéiforme
Chez l'ISP Combo450, une dermatite acnéiforme est survenue chez 4, 0 % des
patients (15/372). Une dermatite acnéiforme de grade 1 ou 2 a été rapportée
chez 3,8 % (14/372) des patients et de grade 3 chez 0,3 % (1/372) des patients.
Aucun événement n'a entraîné un arrêt du traitement. Une modification de la
dose a été rapportée chez 0,5 % (2/372) des patients.
Érythrodysesthésie palmo-plantaire
Une érythrodysesthésie palmo-plantaire peut survenir lorsque le binimetinib
était utilisé en association à l'encorafenib (voir la rubrique Effets
indésirables du RCP de l'encorafenib).
Photosensibilité
Dans l'ISP Combo450, une photosensibilité a été observée chez 4,3 % (16/372)
des patients. La plupart des événements étaient de grade 1 ou 2, un événement
de grade 3 ayant été rapporté chez 0,3 % (1/372) des patients. Aucun événement
n'a conduit à un arrêt définitif traitement. Une interruption ou une
modification de la dose a été rapportée chez 0,3 % (1/372) des patients.
Parésie faciale
Une parésie faciale a été rapportée lorsque le binimetinib était utilisé en
association à l'encorafenib (voir la rubrique Effets indésirables du RCP
de l'encorafenib).
Élévation de la CPK et rhabdomyolyse
Dans
l'ISP Combo450, une élévation de la CPK, généralement peu importante et
asymptomatique, a été rapportée chez 23,9 % des patients (89/372). L'incidence
des effets indésirables de grade 3 ou 4 était de 5,1 % (19/372). Le délai
médian d'apparition du premier événement était de 2,8 mois (allant de 0,5 à 26
mois).
La rhabdomyolyse a été rapportée chez 0,3 % (1/372) des patients traités par le
binimetinib en association à l'encorafenib. Chez ce patient, la rhabdomyolyse a
été observée avec une élévation symptomatique de la CPK de grade 4.
Insuffisance rénale
Des cas d'augmentation de la créatinine sanguine et des cas d'insuffisance
rénale ont été rapportés lors de l'utilisation du binimetinib en association à
l'encorafenib (voir la rubrique Effets indésirables du RCP de
l'encorafenib).
Anomalies biologiques hépatiques
L'incidence des anomalies biologiques hépatiques rapportées dans l'ISP Combo450
est indiquée ci- dessous :
- Augmentation des transaminases : 16,4 % (61/372) tous grades confondus - 6,5 % (24/372) Grade 3
- Augmentation des GGT : 11,3 % (42/372) tous grades confondus - 6,7 % (25/372) Grade 3
Dans l'étude CMEK162B2301, partie 2, bras COMBO300, l'incidence des anomalies biologiques hépatiques est indiquée ci-dessous :
- Augmentation des transaminases : 13,2 % (34/257) tous grades confondus - 5,4 % (14/257) Grade 3-4
- Augmentation des GGT : 14,0 % (36/257) tous grades confondus -4,7 % (12/257) Grade 3-4
Affections gastro-intestinales
Dans l'ISP Combo450, une diarrhée a été rapportée chez 41,7 % des patients
(155/372) et était de grade 3 ou 4 chez 3,8 % des patients (14/372). La
diarrhée a conduit à l'arrêt définitif du traitement chez 0,8 % des patients et
à une interruption ou une modification de dose chez 8,1 % des patients. Une
constipation, de grade 1 ou 2, est survenue chez 24,7 % des patients (92/372).
Une douleur abdominale a été rapportée chez 28,5 % des patients (106/372) et de
grade 3 chez 2,2 % des patients (8/372).
Des nausées sont survenues chez 46 % des patients (171/372) avec un grade 3
observé chez 3,0 % des patients (11/372). Des vomissements sont survenus chez
31,2 % des patients (116/372) avec un grade 3 rapporté chez 1,9 % des patients
(7/372).
Dans l'étude CMEK162B2301, partie 2, bras COMBO300, des nausées sont survenues chez 27,2 % des patients (70/257) avec un grade 3 observé chez 1,6 % des patients (4/257). Des vomissements sont survenus chez 15,2 % des patients (39/257) avec un grade 3 observé chez 0,4 % des patients (1/257). Une diarrhée est survenue chez 28,4 % des patients (73/257) avec un grade 3 observé chez 1,6 % des patients (4/257).
Les troubles gastro-intestinaux ont généralement été traités selon les thérapies usuelles.
Anémie
Dans l'ISP Combo450, une anémie a été rapportée chez 23,1 % des patients
(86/372) ; 7,0 % des patients (26/372) ont présenté des événements de grade 3
ou 4. L'anémie n'a pas entraîné d'arrêt du traitement et a nécessité une
interruption ou modification de dose chez 3,2 % des patients (12/372).
Dans l'étude CMEK162B2301, partie 2, bras COMBO300, une anémie est survenue
chez 9,7 % des patients (25/257) avec un grade 3-4 observé chez 2,7 % des
patients (7/257).
Céphalée
Dans l'ISP Combo450, une céphalée est survenue chez 18,8 % des patients
(70/372) avec un grade 3 observé chez 1,1 % des patients (4/372).
Dans l'étude CMEK162B2301, partie 2, bras COMBO300, une céphalée est survenue chez 12,1 % des patients (31/257) avec un grade 3 observé chez 0,4 % des patients (1/257).
Fatigue
Dans l'ISP Combo450, une fatigue est survenue chez 48,1 % des patients
(179/372) avec un grade 3 ou 4 observé chez 4,3 % des patients (16/372).
Dans l'étude CMEK162B2301, partie 2, bras COMBO300, une fatigue est survenue chez 33,5 % des patients (86/257) avec un grade 3-4 observé chez 1,6 % des patients (4/257).
Populations spéciales
Personnes âgées
Parmi l'ISP Combo450 (n = 372), 230 patients (61,8 %) étaient âgés de moins de 65 ans, 107 patients (28,8 %) étaient âgés de 65 à 74 ans et 35 patients (9,4 %) étaient âgés de plus de 75 ans. De manière globale, aucune différence quant à la sécurité ou à l'efficacité du produit n'a été mise en évidence entre les patients âgés (≥ 65 ans) et les patients plus jeunes à l'exception de la diarrhée et du prurit qui ont été plus fréquemment rapportés chez les patients âgés.
Dans le sous-groupe de patients âgés de ≥ 75 ans, des effets indésirables de grade ≥ 3 (62,9 % versus 47,5 %), des effets indésirables (tous les grades) nécessitant une modification de la dose d'un médicament à l'étude (60,0 % versus 48,1 %) ou conduisant à l'arrêt du traitement (25,7 % versus 7,4%) ont été plus fréquemment rapportés que chez les patients âgés de < 75 ans. Les effets indésirables les plus fréquemment rapportés avec une incidence plus élevée chez les patients âgés de ≥ 75 ans par rapport aux patients âgés de < 75 ans comprenaient la fatigue, les nausées, la diarrhée, les vomissements et l'anémie.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration voir Annexe V.
CONFIRMER avant traitement la présence d'une mutation BRAF V600E évalué par un dispositif médical de diagnostic in vitro (DIV) marqué CE et destiné à l'usage prévu. Si le DIV marqué CE n'est pas disponible, un autre test validé doit être utilisé.
L'efficacité et la sécurité du binimetinib en association avec l'encorafenib ont été établies uniquement chez les patients atteints d'un mélanome porteur des mutations BRAF V600E et V600K, ou atteint d'un CBNPC porteur d'une mutation BRAF V600E. Le binimetinib en association avec l'encorafenib ne doit pas être utilisé chez les patients atteints d'un mélanome ou d'un CBNPC ayant un statut BRAF de type sauvage.SURVEILLANCE du traitement :
- Evaluer la fraction d'éjection ventriculaire gauche par échocardiographie ou
scintigraphie myocardique avant d'initier le binimetinib, un mois après le
début du traitement, puis tous les 3 mois environ ou plus fréquemment au cours
du traitement si cliniquement indiqué.
- A chaque visite, contrôler les symptômes visuels du patient.
- Mesurer les taux de CPK et de créatinine tous les mois pendant les 6 premiers
mois de traitement et en fonction du tableau clinique.
- Mesurer la pression artérielle à l'initiation du traitement et au cours du
traitement, et mettre en place les thérapies usuelles en cas d'hypertension.
- Faire un examen clinique dermatologique avant le début du traitement par
binimetinib, tous les 2 mois pendant le traitement et jusqu'à 6 mois maximum
après l'arrêt de celui-ci. Les lésions suspectes de la peau doivent être
traitées par exérèse cutanée avec un examen dermato-anatomopathologique.
- Faire un examen clinique de la tête et du cou, un scanner thoraco-abdominal
et un examen de la région anale et pelvienne (pour les femmes) et une
numération de la formule sanguine complète avant le début, au cours et à la fin
du traitement, comme cliniquement approprié.
- Mesurer les paramètres biologiques hépatiques avant le début du traitement
par binimetinib, mensuellement pendant les 6 premiers mois de traitement, puis
si cliniquement indiqué.
INFORMER IMMEDIATEMENT un MEDECIN en cas de :
- Sensation de vertige, de fatigue ou d'étourdissement, essoufflement,
avoir l'impression que le cœur bat très fort, rapidement ou
irrégulièrement, gonflement des jambes.
- Maux de tête graves, sensations vertigineuses ou étourdissements ou si la tension artérielle mesurée à la maison à l'aide d'un tensiomètre est beaucoup plus élevée que d'habitude.
- Douleur au niveau de la poitrine, essoufflement soudain ou difficulté à respirer, douleurs dans les jambes avec ou sans gonflement, gonflement des bras et des jambes, un bras ou une jambe froid et pâle
- Vision trouble, perte de la vision ou autres changements de la vision (tels que points colorés dans la vision), halo (voir un contour flou autour des objets), douleurs, gonflement ou rougeur dans l'œil.
- Douleurs, crampes, raideurs ou spasmes musculaires, urine foncée.
- Maux de tête, vertiges ou faiblesse, toux avec crachats de filets de sang ou des caillots de sang, vomi contenant du sang ou qui ressemble à du « marc de café », selles rouges ou noires qui ressemblent à du goudron, présence de sang dans l'urine, douleur à l'estomac (abdominale), saignement vaginal inhabituel.
- Modifications de la peau : nouvelle verrue, peau irritée ou grosseur rougeâtre qui saigne ou ne cicatrise pas ou changement de taille ou de couleur d'un grain de beauté.
- Nausées, essoufflement, rythme cardiaque irrégulier, crampes musculaires, convulsions, trouble des urines, diminution de la production d'urine et fatigue.
FEMMES EN AGE DE PROCREER : utiliser une contraception efficace pendant le traitement par binimetinib et pendant au moins 1 mois après la dernière dose.
OUBLI d'une dose : la dose de binimetinib ne doit pas être prise s'il reste moins de 6 heures avant la prise de la prochaine dose prévue.
VOMISSEMENT : en cas de vomissement peu après la prise du comprimé le patient ne doit pas prendre de dose supplémentaire et prendra la prochaine dose comme initialement prévu.
MAINTENIR une bonne hydratation pendant le traitement.
PRUDENCE en cas de consommation de millepertuis (Hypericum perforatum).
NE PAS CONDUIRE DE VEHICULES OU UTILISER DE MACHINES en cas de troubles de la vision ou de tout autre effet indésirable qui pourrait nuire à l'aptitude à conduire des véhicules ou à utiliser des machines.
- Maux de tête graves, sensations vertigineuses ou étourdissements ou si la tension artérielle mesurée à la maison à l'aide d'un tensiomètre est beaucoup plus élevée que d'habitude.
- Douleur au niveau de la poitrine, essoufflement soudain ou difficulté à respirer, douleurs dans les jambes avec ou sans gonflement, gonflement des bras et des jambes, un bras ou une jambe froid et pâle
- Vision trouble, perte de la vision ou autres changements de la vision (tels que points colorés dans la vision), halo (voir un contour flou autour des objets), douleurs, gonflement ou rougeur dans l'œil.
- Douleurs, crampes, raideurs ou spasmes musculaires, urine foncée.
- Maux de tête, vertiges ou faiblesse, toux avec crachats de filets de sang ou des caillots de sang, vomi contenant du sang ou qui ressemble à du « marc de café », selles rouges ou noires qui ressemblent à du goudron, présence de sang dans l'urine, douleur à l'estomac (abdominale), saignement vaginal inhabituel.
- Modifications de la peau : nouvelle verrue, peau irritée ou grosseur rougeâtre qui saigne ou ne cicatrise pas ou changement de taille ou de couleur d'un grain de beauté.
- Nausées, essoufflement, rythme cardiaque irrégulier, crampes musculaires, convulsions, trouble des urines, diminution de la production d'urine et fatigue.
FEMMES EN AGE DE PROCREER : utiliser une contraception efficace pendant le traitement par binimetinib et pendant au moins 1 mois après la dernière dose.
OUBLI d'une dose : la dose de binimetinib ne doit pas être prise s'il reste moins de 6 heures avant la prise de la prochaine dose prévue.
VOMISSEMENT : en cas de vomissement peu après la prise du comprimé le patient ne doit pas prendre de dose supplémentaire et prendra la prochaine dose comme initialement prévu.
MAINTENIR une bonne hydratation pendant le traitement.
PRUDENCE en cas de consommation de millepertuis (Hypericum perforatum).
NE PAS CONDUIRE DE VEHICULES OU UTILISER DE MACHINES en cas de troubles de la vision ou de tout autre effet indésirable qui pourrait nuire à l'aptitude à conduire des véhicules ou à utiliser des machines.
Femmes en âge de procréer/Contraception chez les femmes
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par binimetinib et pendant au moins 1 mois après la dernière dose.
Grossesse
Il n'existe pas de données sur l'utilisation du binimetinib chez la femme enceinte. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité préclinique). Le binimetinib n'est pas recommandé pendant la grossesse ni chez les femmes en âge de procréer n'utilisant pas de contraception. Si le binimetinib est utilisé pendant la grossesse ou si une grossesse survient pendant le traitement par binimetinib, la patiente devra être informée du danger potentiel pour le fœtus.
Allaitement
Il n'y a pas de données sur l'excrétion du binimetinib ou ses métabolites dans le lait maternel. Un risque pour les nouveau-nés/nourrissons allaités ne peut pas être exclu. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre le traitement avec Mektovi en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la mère.
Fertilité
Aucune donnée n'est disponible sur les effets de binimetinib sur la fertilité chez les humains.
Effets d'autres médicaments sur le binimetinib
Le binimetinib est principalement métabolisé via glycurono-conjugaison médiée par UGT1A1. Il est peu probable que l'étendue des interactions médicamenteuses médiées par l'UGT1A1 soit cliniquement pertinente (voir rubrique Propriétés pharmacocinétiques) ; toutefois, comme cela n'a pas été évalué dans une étude clinique formelle, les inducteurs de l'UGT1A1 (comme la rifampicine et le phénobarbital) et les inhibiteurs (comme indinavir, atazanavir, sorafénib) doivent être administrés avec prudence.
Alors que l'encorafenib est un inhibiteur relativement puissant et réversible de l'UGT1A1, aucune différence au niveau de l'exposition au binimetinib n'a été observée du point de vue clinique lors de l'administration concomitante du binimetinib et de l'encorafenib (voir rubrique Propriétés pharmacocinétiques).
Les inducteurs des enzymes CYP1A2 (tels que la carbamazépine et la rifampicine) et les inducteurs du transport de la Pgp (tels que le millepertuis ou la phénytoïne) peuvent réduire l'exposition au binimetinib, ce qui pourrait diminuer son efficacité.
Effets du binimetinib sur d'autres médicaments
Le binimetinib est un inducteur potentiel de CYP1A2 et il convient de faire preuve de prudence lorsqu'il est associé à des substrats sensibles (tels que la duloxétine ou la théophylline).
Le binimetinib est un inhibiteur faible de l'OAT3, et il convient de faire preuve de prudence lorsqu'il est associé à des substrats sensibles (tels que la pravastatine ou la ciprofloxacine).
Le traitement par le binimetinib en association avec l'encorafenib doit être initié et supervisé par un médecin expérimenté dans l'utilisation de médicaments anticancéreux.
Test de mutation BRAF
Avant toute administration du binimetinib en association avec l'encorafenib, les patients doivent avoir la confirmation de la présence d'une mutation BRAF V600E évalué par un dispositif médical de diagnostic in vitro (DIV) marqué CE et destiné à l'usage prévu. Si le DIV marqué CE n'est pas disponible, un autre test validé doit être utilisé.
L'efficacité et la sécurité du binimetinib en association avec l'encorafenib ont été établies uniquement chez les patients atteints d'un mélanome porteur des mutations BRAF V600E et V600K, ou atteint d'un CBNPC porteur d'une mutation BRAF V600E. Le binimetinib en association avec l'encorafenib ne doit pas être utilisé chez les patients atteints d'un mélanome ou d'un CBNPC ayant un statut BRAF de type sauvage.
Posologie
La dose recommandée de binimetinib est de 45 mg (3 comprimés de 15 mg ou 1 comprimé de 45 mg) deux fois par jour à environ 12 heures d'intervalle, correspondant à une dose quotidienne totale de 90 mg.
Adaptation posologique
La prise en charge des effets indésirables peut nécessiter une réduction de dose, une interruption temporaire ou un arrêt définitif du traitement (voir Tableaux 1 et 2 ci-dessous).
Chez les patients recevant 45 mg de binimetinib deux fois par jour, la dose réduite recommandée de binimetinib est de 30 mg deux fois par jour. Une réduction de dose inférieure à 30 mg deux fois par jour n'est pas recommandée. Le traitement doit être arrêté si le patient n'est pas en mesure de tolérer 30 mg deux fois par jour.
Si l'effet indésirable à l'origine d'une réduction de dose est correctement pris en charge, une ré- augmentation de dose à 45 mg deux fois par jour peut être envisagée. Il n'est pas recommandé de revenir à la dose à 45 mg deux fois par jour si la réduction de la dose était due à une dysfonction ventriculaire gauche (DVG) ou à une toxicité de grade 4.
Les recommandations d'adaptation posologique en cas d'effets indésirables sont présentées ci-dessous et dans les Tableaux 1 et 2.
Si des toxicités liées au traitement surviennent lorsque le binimetinib est utilisé en association avec l'encorafenib, les doses des deux traitements doivent simultanément être réduites, interrompues ou arrêtées. Les exceptions nécessitant des réductions de dose uniquement pour l'encorafenib (effets indésirables principalement liés à l'encorafenib) sont : l'érythrodysesthésie palmo-plantaire (EPP), l'uvéite y compris l'iritis et l'iridocyclite et l'allongement de l'intervalle QTc.
Si l'une de ces toxicités se produit, veuillez consulter la rubrique Posologie et mode d'administration du résumé des caractéristiques du produit (RCP) de l'encorafenib pour les instructions de modification de la dose pour l'encorafenib.
Si le traitement par le binimetinib est temporairement interrompu, la dose d'encorafenib doit être réduite à 300 mg une fois par jour pendant l'interruption du traitement par le binimetinib (voir Tableaux 1 et 2) compte-tenu de la moins bonne tolérance du traitement par encorafenib pris seul à la dose de 450 mg. Si le binimetinib est définitivement arrêté, l'encorafenib doit être interrompu.
Si
l'encorafenib est temporairement interrompu (voir la
rubrique Posologie et mode d'administration du RCP de l'encorafenib), le binimetinib doit
être interrompu. Si l'encorafenib est définitivement
arrêté, le binimetinib doit également être
définitivement arrêté.
Pour plus d'informations sur la posologie et les modifications de dose
recommandées pour l'encorafenib, voir la rubrique Posologie
et mode d'administration du RCP de l'encorafenib.
Tableau 1 : Adaptations posologiques recommandées pour le binimetinib (utilisé en association à l'encorafenib) lors de la survenue de certains effets indésirables
| Sévérité de l'effet indésirablea | Binimetinib |
| Réactions cutanées | |
|
Le
binimetinib doit être maintenu. Si l'éruption cutanée s'aggrave ou ne s'améliore pas au bout de 2 semaines de traitement, le binimetinib doit être interrompu jusqu'à une amélioration au grade 0 ou 1, puis réintroduit à la même dose s'il s'agit de la première survenue ou réintroduit à une dose réduite s'il s'agit d'une récidive de grade 2. |
|
Le binimetinib doit être interrompu jusqu'à une amélioration à un grade 0 ou 1 et réintroduit à la même dose s'il s'agit de la première survenue ou alors réintroduit à une dose réduite s'il s'agit d'une récidive de grade 3. |
|
Le binimetinib doit être définitivement arrêté. |
| Atteintes oculaires | |
|
Le
binimetinib doit être interrompu jusqu'à 2 semaines
et le contrôle ophtalmologique doit être répété, y compris l'acuité visuelle.
|
|
Le binimetinib doit être définitivement arrêté. |
|
Le binimetinib doit être définitivement arrêté. |
| Événements cardiaques | |
|
La
FEVG doit être évaluée toutes les 2 semaines.
Le
binimetinib doit être interrompu pendant un maximum
de 4 semaines. Le binimetinib doit être réintroduit
à une dose réduite si tous les éléments suivants sont présents dans les 4
semaines :
|
|
Le
binimetinib doit être définitivement arrêté. La FEVG doit être évaluée toutes les 2 semaines jusqu'à résolution. |
| Rhabdomyolyse/Elévation des taux de créatine phosphokinase (CPK) | |
|
La dose de binimetinib doit être maintenue et s'assurer que le patient est correctement hydraté. |
|
Le binimetinib doit être interrompu jusqu'à une amélioration au grade 0 ou 1. S'assurer de bonne hydratation du patient. |
|
Le
binimetinib doit être interrompu jusqu'à une
amélioration au grade 0 ou 1.
|
| Maladie thromboembolique veineuse (MTEV) | |
|
Le
binimetinib doit être interrompu.
|
|
Le binimetinib doit être définitivement arrêté. |
| Anomalies biologiques hépatiques | |
|
La
dose de binimetinib doit être maintenue.
|
|
Le
binimetinib doit être interrompu pendant 4 semaines
au maximum.
|
|
Le
binimetinib doit être interrompu pendant 4 semaines
au maximum.
|
|
L'arrêt définitif de binimetinib doit être envisagé. |
|
Le binimetinib doit être définitivement arrêté. |
| Pneumopathie interstitielle diffuse (PID)/pneumopathie inflammatoire | |
|
Le
binimetinib doit être interrompu pendant 4 semaines
au maximum.
|
|
Le binimetinib doit être définitivement arrêté. |
a Critères communs de terminologie de l'institut national
contre le cancer pour les événements indésirables (« National Cancer Institute
Common TerminologyCriteria for Adverse Events », NCI
CTCAE) version 4.03
Tableau 2 : Adaptations posologiques recommandées pour le binimetinib (utilisé en association avec l'encorafenib) lors de la survenue d'autres effets indésirables
| Sévérité des effets indésirables | Binimetinib |
|
Le
binimetinib doit être interrompu pendant 4 semaines
au maximum.
|
|
Le
binimetinib doit être interrompu pendant 4 semaines
au maximum.
Ou, le binimetinib doit être définitivement arrêté. |
|
|
|
|
Durée du traitement
Il
convient de continuer le traitement jusqu'à ce qu'il n'y ait plus de bénéfice
pour le patient ou en cas de survenue de toxicité inacceptable.
Omissions de doses
En
cas d'oubli d'une dose de binimetinib, la dose de binimetinib ne doit pas être prise s'il reste moins de 6
heures avant la prise de la prochaine dose prévue.
Vomissement
En
cas de vomissement après l'administration de binimetinib,
le patient ne doit pas prendre de dose supplémentaire et prendra la prochaine
dose comme initialement prévu.
Populations spéciales
Personnes âgées
Aucune
adaptation posologique n'est nécessaire pour les patients âgés de 65 ans et
plus (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucun
ajustement de dose n'est nécessaire chez les patients présentant une insuffisance
hépatique légère (Child-Pugh A).
L'encorafenib étant déconseillé chez les patients atteints d'une insuffisance hépatique modérée (Child- Pugh B) ou sévère (Child-Pugh C), l'administration de binimetinib n'est pas recommandée dans ce cas (voir la rubrique Posologie et mode d'administration du RCP de l'encorafenib).
Insuffisance rénale
Aucun
ajustement posologique n'est nécessaire chez les patients présentant une
insuffisance rénale (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La
sécurité et l'efficacité de binimetinib n'ont pas
encore été établies chez les enfants et les adolescents. Aucune donnée n'est
disponible.
Mode d'administration
Mektovi s'administre par voie orale.
Les
comprimés doivent être avalés entiers avec de l'eau. Ils peuvent être pris
pendant ou en dehors des repas.
Durée de conservation :
3 ans.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
La dose la plus élevée de binimetinib administré seul, évaluée dans les études cliniques était de 80 mg deux fois par jour par voie orale et associée à des toxicités oculaire (chorio-rétinopathie) et cutanée (dermatite acnéiforme).
Il
n'y a pas d'antidote en cas de surdosage. En cas de surdosage, le
patient doit être pris en charge avec des soins de support et une
surveillance appropriée si nécessaire.
Le binimetinib étant lié aux protéines plasmatiques, une hémodialyse
serait probablement inefficace dans le traitement d'un surdosage par
binimetinib.
Classe pharmacothérapeutique : agents antinéoplasiques, inhibiteurs de la protéine kinase, Code ATC : L01EE03
Mécanisme d'action
Le binimetinib est un inhibiteur non compétitif réversible de l'activation du signal régulé par MEK 1 (mitogen-activated extracellular signal regulated kinase 1) et MEK2. Dans un système acellulaire, le binimetinib inhibe MEK1 et MEK2 avec une concentration inhibitrice 50 % (CI50) de l'ordre de 12 à 46 nM. Les protéines MEK sont des composants de la voie régulée par la kinase ERK (extracellular signal-related kinase) qui favorise la prolifération cellulaire. Dans le mélanome et d'autres cancers, cette voie est souvent activée par des formes mutées de BRAF qui activent MEK. Le binimetinib inhibe l'activation de MEK par BRAF et inhibe l'activité de la kinase MEK. Le binimetinib inhibe la prolifération des lignées cellulaires de mélanome exprimant la mutation BRAF V600 et démontre des effets anti-tumoraux dans les modèles animaux porteurs d'un mélanome avec une mutation BRAF V600.
Association à l'encorafenib
Le binimetinib et l'encorafenib (inhibiteur de BRAF, voir la rubrique Propriétés pharmacodynamiques du RCP de l'encorafenib) inhibent tous les deux la voie MAPK et conduisant à une activité anti-tumorale plus élevée que le traitement en monothérapie, par l'un ou l'autre médicament.
Efficacité et sécurité cliniques
Mélanome inopérable ou métastatique présentant
une mutation BRAF V600
La sécurité et l'efficacité de binimetinib associé à
l'encorafenib ont été évaluées dans une étude de
phase III randomisée (1:1:1) en deux parties, multicentrique, ouverte et
contrôlée par traitement actif, chez des patients atteints d'un mélanome
inopérable ou métastatique présentant une mutation BRAF V600 E ou K (étude
CMEK162B2301) mise en évidence à l'aide d'un test de détection des mutations de
BRAF. Les patients présentaient un mélanome primitif histologiquement confirmé
cutané ou inconnu mais ceux qui étaient atteints d'un mélanome choroïdien ou
muqueux ont été exclus. Les patients étaient autorisés à recevoir un traitement
adjuvant préalable et une ligne préalable d'immunothérapie pour une maladie
localement avancée inopérable ou métastatique. Un traitement préalable par des
inhibiteurs de BRAF/MEK n'était pas autorisé.
Étude CMEK162B2301, partie 1
Dans la partie 1, les patients de l'étude ont été randomisés pour recevoir le binimetinib 45 mg par voie orale deux fois par jour plus encorafenib 450 mg par voie orale une fois par jour
(COMBO450, n = 192), l'encorafenib 300 mg par voie
orale une fois par jour (ci-après dénommé Enco 300, n
= 194) ou le vemurafenib 960 mg par voie orale deux
fois par jour (ci-après dénommé Vem, n = 191). Le
traitement a été poursuivi jusqu'à progression de la maladie ou une toxicité
inacceptable. La randomisation a été stratifiée selon les stades (IIIB, IIIC,
IVM1a ou IVM1b, vs IVM1c) de la classification de l'AJCC (American Joint Committee on Cancer) et l'indice de performance ECOG (Eastern Cooperative Oncology Group) (0 vs 1) et selon qu'une immunothérapie
préalable a été reçue ou non pour la maladie inopérable ou métastatique.
Le critère d'évaluation principal en termes d'efficacité était la survie sans progression (SSP) du bras COMBO450 comparé au vemurafenib évalué par un comité indépendant de revue centralisée (BIRC, Blinded Independent Review Committee). La survie sans progression évaluée par les investigateurs (évaluation des investigateurs) était une analyse complémentaire de l'analyse du critère principal. Un critère d'évaluation secondaire supplémentaire comprenait la survie sans progression de COMBO450 comparé à Enco 300. D'autres comparaisons secondaires de l'efficacité entre COMBO450 et le vemurafenib ou l'Enco 300 comprenaient la survie globale (SG), le taux de réponse objective (ORR), la durée de la réponse (DR) et le taux de contrôle de la maladie (TCM) comme évalués par le BIRC et par l'évaluation des investigateurs.
L'âge médian des patients était de 56 ans (de 20 à 89 ans), 58 % étaient des hommes, 90 % de type caucasien et 72 % des patients présentaient un indice de performance ECOG de 0 à l'inclusion. La plupart des patients étaient atteints d'une maladie métastatique (95 %) et étaient de Stade IVM1c (64 %) ; 27 % des patients présentaient à l'inclusion un taux élevé de lactate déshydrogénase sérique (LDH) et 45 % des patients avaient au moins 3 organes présentant des lésions tumorales à l'inclusion et 3,5 % avaient des métastases cérébrales. 27 patients (5 %) avaient préalablement reçu des inhibiteurs de « points de contrôle » (anti-PD1/PDL1 ou ipilimumab [anti-CTL-A4]) (8 patients dans le bras COMBO450 (4 %) ; 7 patients dans le bras vemurafenib (4 %) ; 12 patients dans le bras Enco 300 (6 %)),dont 22 patients dans un cadre métastatique (6 patients dans le bras COMBO450 ; 5 patients dans le bras vemurafenib ; 11 patients dans le bras Enco 300) et 5 patients dans le cadre d'un traitement adjuvant (2 patients dans le bras COMBO450 ; 2 patients dans le bras vemurafenib ; 1 patient dans le bras Enco 300).
La durée médiane d'exposition était de 11,7 mois chez les patients traités par COMBO450, de 7,1 mois chez les patients traités par Enco 300 et de 6,2 mois chez les patients traités par vemurafenib. La dose- intensité relative (RDI) médiane pour COMBO450 était de 99,6 % pour le binimetinib et de 100 % pour l'encorafenib ; la RDI médiane était de 86,2 % pour Enco 300 et de 94,5 % pour le vemurafenib.
La partie 1 de l'étude CMEK162B2301 a démontré une amélioration statistiquement significative de la survie sans progression chez les patients traités par COMBO450 par rapport à ceux traités par vemurafenib. Le tableau 4 résume la survie sans progression et les autres résultats d'efficacité basés sur la revue faite en aveugle par un comité indépendant de radiologues.
Les résultats d'efficacité reposant sur l'évaluation des investigateurs étaient cohérents avec l'évaluation centrale indépendante. Des analyses de sous-groupes en dehors de toute stratification ont démontré des estimations ponctuelles en faveur du COMBO450, notamment le taux de LDH à l'inclusion, l'indice de performance ECOG et le stade AJCC.
Tableau 4 : Étude CMEK162B2301, partie 1 : résultats de la survie sans progression et de la réponse globale confirmée (examen central indépendant)
|
Encorafenib
+ binimetinib n = 192 (COMBO450) |
Encorafenib
n = 194 (Enco 300) |
Vemurafenib
n = 191 (Vem) |
|
| Cut-off au 19 mai 2016 | |||
| Survie Sans Progression (analyse principale) | |||
| Nombre d'événements (%) | 98 (51,0) | 96 (49,5) | 106 (55,5) |
|
Médiane,
en mois (IC à 95 %) |
14,9
(11,0 ; 18,5) |
9,6
(7,5 ;14,8) |
7,3
(5,6 ; 8,2) |
|
Hazard
Ratioa (IC à 95 %) (par rapport à Vem) Valeur p (test Mantel-Haenzel stratifié)b |
0,54
(0,41 ; 0,71) < 0,0001 |
||
|
Hazard
Ratioa (IC à 95 %) (par rapport à Vem) Valeur nominale de p |
0,68
(0,52 ; 0,90) 0,007 |
||
|
Hazard
Ratioa (IC à 95 %) (par rapport à Enco 300) Valeur p (test Mantel-Haenzel stratifié)b |
0,75
(0,56 ; 1,00) 0,051 |
||
| Réponses globales confirmées | |||
|
Taux
de réponse globale, n (%) (IC à 95 %) |
121
(63,0) (55,8 ; 69,9) |
98
(50,5) (43,3 ; 57,8) |
77
(40,3) (33,3 ; 47,6) |
| Réponse complète, n (%) | 15 (7,8) | 10 (5,2) | 11 (5,8) |
| Réponse partielle, n (%) | 106 (55,2) | 88 (45,4) | 66 (34,6) |
| Maladie stable, n (%) | 46 (24,0) | 53 (27,3) | 73 (38,2) |
|
Taux
de contrôle de la maladie n (%) (IC à 95 %) |
177
(92,2) (87,4 ; 95,6) |
163
(84,0) (78,1 ; 88,9) |
156
(81,7) (75,4 ; 86,9) |
| Durée de réponse | |||
|
Médiane,
en mois (IC à 95 %) |
16,6
(12,2 ; 20,4) |
14,9
(11,1 à Non estimable) |
12,3
(6,9 ; 16,9) |
IC =
Intervalle de confiance ; Taux de contrôle de la maladie (Réponse Complète +
Réponse Partielle + Maladie stable + Non-Réponse Complète/Non-Progression de la
maladie ; Non-Réponse complète/Non-Progression de la maladie s'applique
uniquement aux patients sans lésion cible qui n'ont pas atteint la Réponse
Complète ou n'ont pas une Progression de la maladie).
Vem = vemurafenib.
HR = rapport des risques instantanés (Hazard Ratio)
a Rapport des risques instantanés selon un modèle à risques proportionnels de Cox stratifié
b Valeur p du test Mantel-Haenzel (test bilatéral)
Vem = vemurafenib.
HR = rapport des risques instantanés (Hazard Ratio)
a Rapport des risques instantanés selon un modèle à risques proportionnels de Cox stratifié
b Valeur p du test Mantel-Haenzel (test bilatéral)
Qualité de vie (QdV) cut-off au 19 mai 2016)
Le score FACT-M (Functional Assessment
of Cancer Therapy-Melanoma), le questionnaire de
qualité de vie de l'Organisation européenne pour la recherche et le traitement
du cancer (EORTC QLQ-C30) et le questionnaire EQ-5D-5L (EuroQoL-5 Dimension-5 Level) ont été utilisés pour explorer les mesures
rapportées par les patients, concernant la qualité de vie liée à la santé, les
critères fonctionnels, les symptômes liés au mélanome et les évènements
indésirables liés au traitement Une détérioration définitive de 10 % dans le
FACT-M et dans l'EORTC QLQ-C30 a été considérablement retardée chez les
patients traités par COMBO450 par rapport aux autres traitements. Le temps
médian jusqu'à détérioration définitive de 10 % dans le score FACT-M n'a pas
été atteint dans le bras COMBO450 et était de 22,1 mois (IC à 95 % : 15,2 non
estimable) dans le bras vemurafenib avec un Hazard Ratio
pour la différence de 0,46 (IC à 95 % : 0,29 à 0,72). Une analyse du délai
jusqu'à une détérioration définitive de 10 % dans le score EORTC QLQ-C30 a
donné des résultats semblables.
Les
patients recevant COMBO450 n'ont signalé aucun changement ou une légère
amélioration du changement moyen par rapport à l'indice EQ-5D-5L à l'inclusion
à toutes les visites, alors que les patients recevant vemurafenib
ou encorafenib ont signalé des diminutions à toutes
les visites (avec des différences significatives sur le plan statistique). Une
évaluation du changement de score au fil du temps a donné la même tendance pour
l'EORTC QLQ-C30 et à toutes les visites pour FACT-M.
Étude CMEK162B2301, partie 2
La partie 2 de l'étude CMEK162B2301 était conçue pour évaluer la contribution
du binimetinib dans le cadre de l'association de l'encorafenib au binimetinib.
La
survie sans progression de l'encorafenib à 300 mg par
voie orale une fois par jour en association au binimetinib
à 45 mg administré par voie orale deux fois par jour (COMBO300, n = 258) a été
comparée à la survie sans progression d'Enco 300 (n =
280, dont 194 patients issus de la partie 1 et 86 patients de la partie 2). Le
recrutement dans la partie 2 a commencé après la randomisation de tous les
patients de la partie 1.
Analyse finale de l'efficacité de l'étude CMEK162B2301, parties 1 et 2 (cut-off au 31 mars 2023)
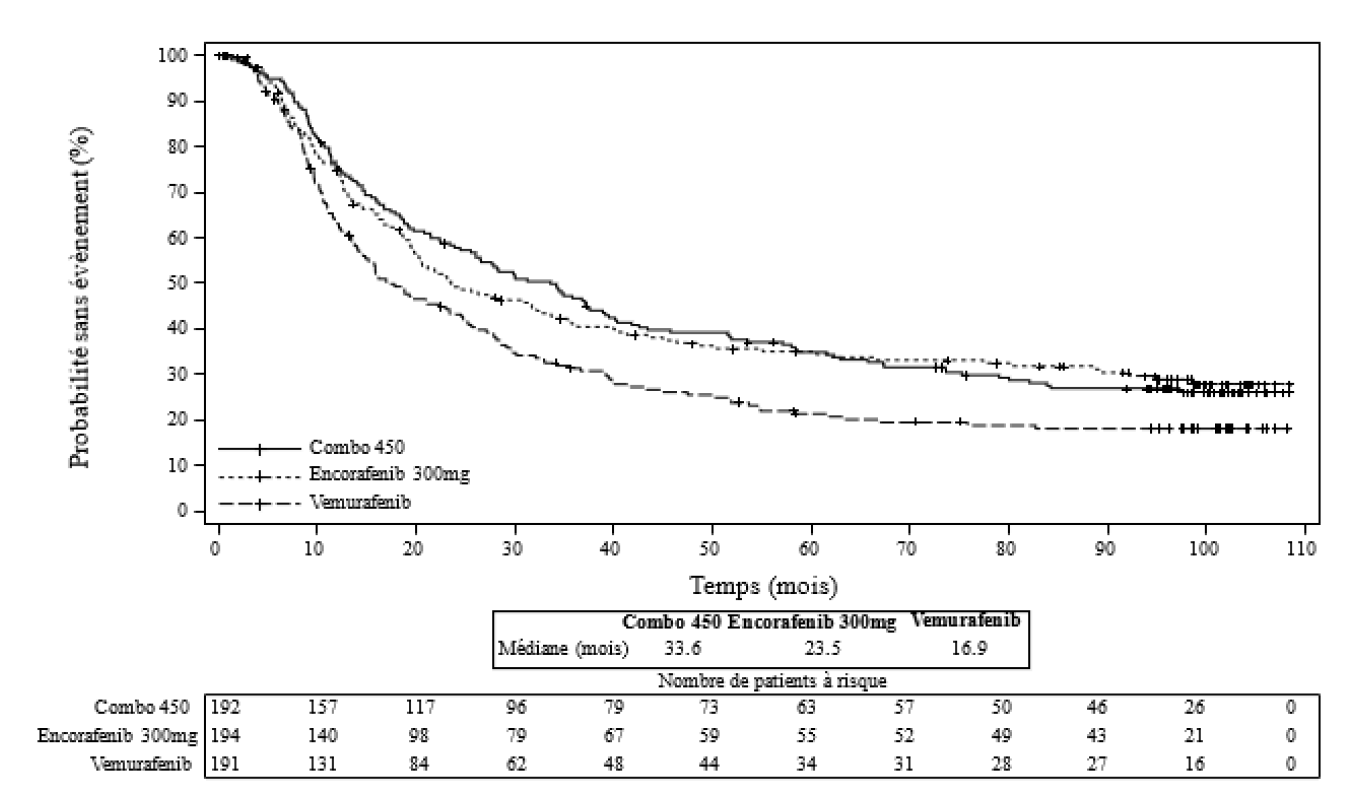
L'analyse finale de l'efficacité était cohérente avec les résultats de l'analyse intermédiaire et a montré un bénéfice en termes de survie globale pour Combo 450 par rapport au vemurafenib Hazard Ratio (HR) 0,67 [95% IC: 0.53,0.84] avec une survie globale médiane de 33,6 mois contre 16,9 mois). Les résultats de la SSP et du TRO_TRG (selon le BIRC) ont également confirmé un avantage numérique en faveur de Combo 450, avec une SSP médiane de 7,6 mois plus longue dans le groupe Combo 450 par rapport au groupe vemurafenib, voir tous les résultats d'efficacité finaux détaillés dans le tableau 5 et les figures 1 et 2 ci-dessous.
De plus, l'analyse finale de la partie 2 a montré une différence numérique en terme de SG pour Combo 300 (partie 2) par rapport à la monothérapie Enco 300 (parties 1+2) (HR 0,89 [IC à 95 % : 0,72, 1,09] avec une SG médiane de 27,1 mois [IC à 95 % : 21,6-33,3] contre 22,7 mois [IC à 95 % : 19,3-29,3]). La SSP médiane est restée plus longue dans le bras Combo 300 (partie 2) que dans le groupe Enco 300 (parties 1+2) avec des estimations de SSP médianes de 12,9 mois (IC à 95 % : 10,9, 14,9) et 9,2 mois (IC à 95 % : 7,4, 11,1), respectivement. Le taux de réponse objective confirmé (selon le BIRC) était de 67,8 % (IC à 95 % : 61,8, 73,5) et de 51,4 % (IC à 95 % : 45,4, 57,4) dans les groupes Combo 300 (partie 2) et Enco 300 (parties 1 + 2), respectivement. Des résultats similaires ont été observés lors de l'évaluation de l'investigateur.
Tableau 5 : Étude CMEK162B2301 : Résultats définitifs sur SSP, SG et TRO confirmés (cut-off au 31 mars 2023)
|
Encorafenib + binimetinib N=192 (Combo 450) |
Encorafenib
N=194 (Enco 300) |
Vemurafenib
N=191 (Vem) |
|
| Analyse finale, cut-off au du 31 Mars 2023 | |||
| SSP (selon le BIRC) | |||
| Nombre d'événements (%) | 123 (64.1) | 119 (61.3) | 121 (63.4) |
|
Médiane
a, en mois (IC à 95 %) |
14.9
(11.0, 20.2) |
9.6
(7.4, 14.8) |
7.3
(5.6, 7.9) |
|
Hazard
Ratioc (IC à 95 %) (par rapport à Vem) Valeur p du log-rank (unilatéral)* |
0.51
(0.39, 0.66) <0.0001 |
0.68
(0.53, 0.88) 0.0017 |
|
|
Hazard
Ratioc (IC à 95 %) (par rapport à Enco300) Valeur p du log-rank (unilatéral)* |
0.77
(0.60, 0.99) 0.0214 |
||
| Survie globale | |||
| Nombre d'événements (%) | 139 (72.4) | 125 (64.4) | 147 (77.0) |
|
Médiane
a, en mois (IC à 95 %) |
33.6
(24.4, 39.2) |
23.5
(19.6, 33.6) |
16.9
(14.0, 24.5) |
|
Probabilité
de survie b à 1 an % (95 % IC) |
75.5 (68.8, 81.0) |
74.6 (67.6, 80.3) |
63.1 (55.7, 69.7) |
| à 2 ans % (95 % IC) | 57.7 (50.3, 64.3) | 49.1 (41.5, 56.2) | 43.2 (35.9, 50.2) |
| à 3 ans % (95 % IC) | 46.5 (39.3, 53.4) | 40.9 (33.6, 48.1) | 31.4 (24.8, 38.2) |
| à 5 ans % (95 % IC) | 34.7 (28.0, 41.5) | 34.9 (27.9, 42.0) | 21.4 (15.7, 27.8) |
| à 9 ans % (95 % IC) | 26.0 (19.8, 32.5) | 27.8 (21.1, 34.8) | 18.2 (12.8, 24.3) |
|
HRc (9 5% IC) (vs Vem) Valeur p du log-rank (unilatéral)* |
0.67
(0.53, 0.84) 0.0003 |
0.74
(0.58, 0.94) 0.0063 |
|
|
HRc (95 % IC) (vs Enco 300) Valeur p du log-rank (unilatéral)* |
0.93
(0.73, 1.19) 0.2821 |
||
| Réponse globale confirmée optimale (selon le BIRC) | |||
|
TRO
confirmée d, n (%) (95 % IC) |
123
(64.1) (56.8, 70.8) |
100
(51.5) (44.3, 58.8) |
78
(40.8) (33.8, 48.2) |
| RC, n (%) | 29 (15.1) | 17 (8.8) | 16 (8.4) |
| RP, n (%) | 94 (49.0) | 83 (42.8) | 62 (32.5) |
| MS, n (%) | 44 (22.9) | 52 (26.8) | 71 (37.2) |
|
TCM
d, n (%) (95 % IC) |
177
(92.2) (87.4, 95.6) |
163
(84.0) (78.1, 88.9) |
155
(81.2) (74.8, 86.4) |
| Durée de la réponse (selon le BIRC) | |||
|
Médiane,
mois (95 % IC) |
18.6
(12.7, 27.6) |
15.5
(11.1, 29.5) |
12.3
(6.9, 14.5) |
|
IC = intervalle de confiance ; RC = réponse complète ; RP
= réponse partielle ; MS = maladie stable ; TCM : taux de contrôle de la maladie (RC+RP+MS+Non- RC/Non-Progression de la maladie; HR : hazard ratio ; TRO : taux de réponse objective (RC+RP). La RP et la CR sont confirmées par des évaluations répétées effectuées au moins 4 semaines après que les critères de réponse ont été établis pour la première fois. a La médiane (temps jusqu'à l'évènement) et ses IC à 95 % sont générés par l'estimation KM avec la méthode Broomeyer et Crowley b Probabilité de survie (obtenues à partir des estimations de survie KM, formule de Greenwood utilisée pour les IC) c Le test du log-rank et le modèle Cox PH sont stratifiés par stade IVRS AJCC et statut de performance ECOG. d Les IC estimés à 95 % sont obtenus à l'aide de la méthode exacte Clopper-Pearson *valeur p nominale |
|||
Figure 1 Étude CMEK162B2301 : Diagramme de Kaplan-Meier de SSP par BIRC (cut-off au 31 mars 2023)
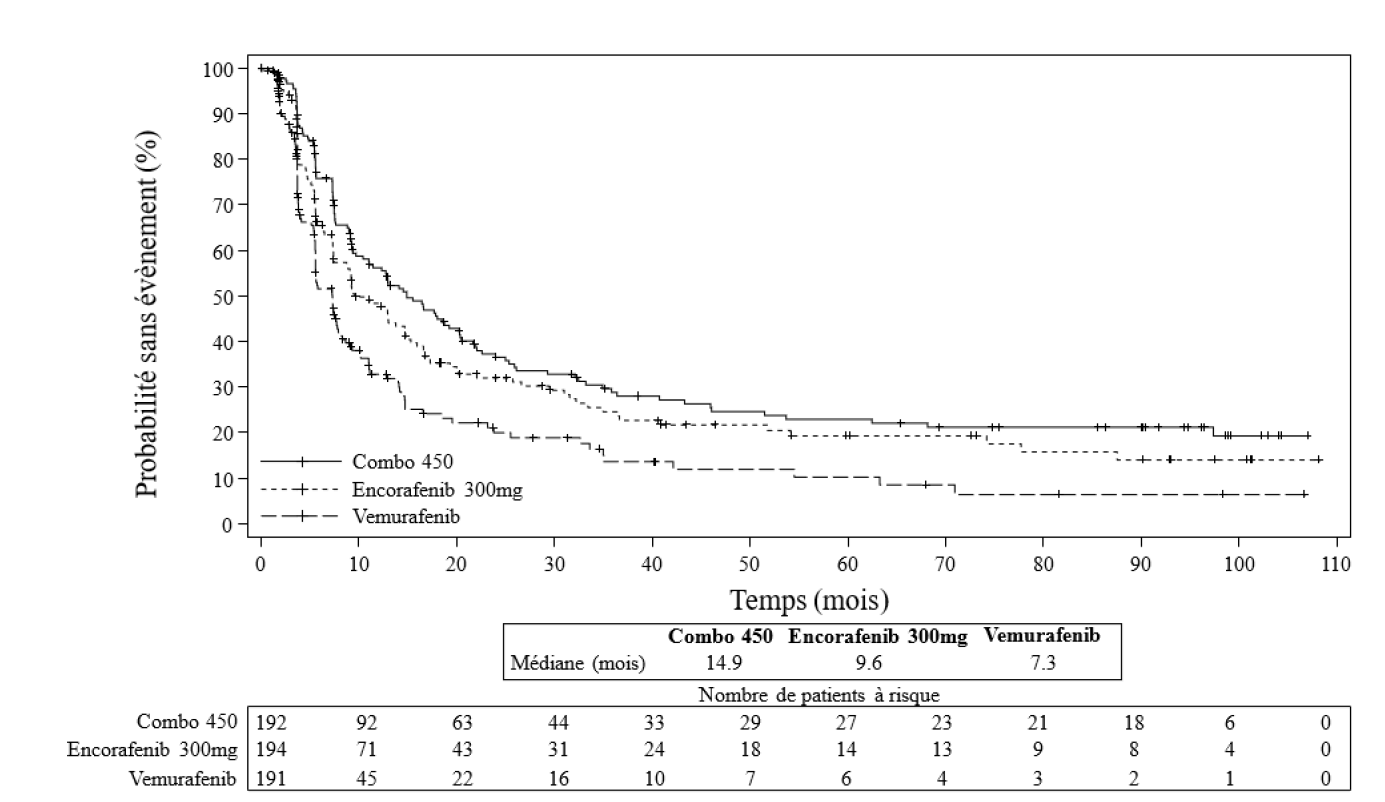
Figure 2 Etude CMEK162B2301 : Diagramme de Kaplan-Meier de la survie globale (cut-off au 31 Mars 2023)
Cancer
bronchopulmonaire non à petites cellules muté BRAF
V600E - Étude ARRAY-818-202
La sécurité et l'efficacité du binimetinib en
association avec l'encorafenib ont été étudiées dans
le cadre d'une étude de phase II, ouverte, multicentrique et non comparative
(étude ARRAY-818-202, PHAROS). Les patients devaient avoir un CBNPC
métastatique confirmé histologiquement avec une mutation BRAF V600E, un indice
de performance ECOG de 0 ou 1 et une maladie mesurable.
Les patients étaient naïfs de traitement ou avaient reçu 1 ligne de traitement
systémique antérieur dans le cadre de leur maladie métastatique.
L'utilisation antérieure d'inhibiteurs de BRAF ou d'inhibiteurs de MEK était
interdite.
Les patients ont été recrutés sur la base de la détermination d'une mutation BRAF V600E dans le tissu tumoral ou le sang (par exemple, test génétique ADNct) par un test de laboratoire local. La confirmation centrale du statut de mutation BRAF V600E (c'est-à-dire toute variant court avec un effet protéique V600E) a été réalisée sur des tissus tumoraux frais ou d'archives prélevés lors de l'inscription et a utilisé le test FoundationOne CDx - F1CDx (tissus).
La sensibilité analytique a été évaluée par l'étude de limite de détection (LoD) pour F1CDx en utilisant la méthode du taux de réussite (définie comme le niveau le plus bas avec une détection ≥ 95 %) en évaluant la fréquence allélique variable (FAV) pour les variantes courts. Pour F1CDx, la LoD médiane de substitution a été déterminée à 3,2 % de FAV.
Au total, 98 patients ont été inclus et traités par binimetinib 45 mg par voie orale deux fois par jour et par encorafenib 450 mg par voie orale une fois par jour. Le traitement s'est poursuivi jusqu'à progression de la maladie ou toxicité inacceptable.
Le principal critère d'évaluation de l'efficacité était le taux de réponse objective (TRO) selon RECIST v1.1, tel qu'évalué par une revue radiologique indépendante (IRR). Les critères d'évaluation secondaires comprenaient la durée de réponse (DoR), le taux de contrôle de la maladie (TCM), la survie sans 26 progression (SSP) et la survie globale (SG). Les résultats de l'analyse primaire avec 18,2 mois pour les patients naïfs de traitement et 12,8 mois pour les patients pré-traités sont présentés ci-dessous.
Sur les 98 patients inclus dans cette étude, 59 (60,2 %) étaient naïfs de traitement. L'âge médian des patients était de 70 ans (47-86), 53 % étaient des femmes, 88 % étaient blancs et 30 % n'avaient jamais fumé. 74 % avaient un indice de performance (PS) ECOG de 1 à l'inclusion (67,8 % des participants avaient un PS 1 à l'inclusion dans la population naïve de traitement et 82,1 % dans la population pré-traitée). Tous les patients présentaient une maladie métastatique, parmi lesquels 8 % avaient des métastases cérébrales au diagnostic et 97 % avaient un adénocarcinome.
Au
moment de l'analyse principale, la durée médiane d'exposition était de 15,1
mois chez les patients naïfs de traitement et de 5,4 mois chez les patients
préalablement traités. Dans la population globale, l'intensité de dose relative
(IDR) médiane était de 95,4 % pour le binimetinib et
de 99,2 % pour l'encorafenib.
Au moment de l'analyse principale, le critère d'évaluation principal du TRO
évalué par IRR dans la population naïve de traitement était de 74,6 % (IC à 95
% : 61,6 ; 85,0), dont 9 (15,3 %) réponse complète (RC) et 35 (59,3 %) réponse
partielle (RP) .
Le TRO par IRR dans la population pré-traitée était de 46,2 % (IC à 95 % : 30,1, 62,8), dont 4 (10,3 %) RC et 14 (35,9 %) RP.
Les résultats mis à jour avec un suivi supplémentaire de 10 mois (durée médiane d'exposition de 16,3 mois chez les patients naïfs de traitement et de 5,5 mois chez les patients préalablement traités) sont présentés dans le Tableau 6.
Tableau 6 : Etude ARRAY-818-202 : Résultats d'efficacité
| Binimetinib avec encorafenib | ||
|
Naïf de traitement
(N = 59) |
Pré-traités
(N = 39) |
|
| TRO par IRR | ||
| TRO, % (95 % IC) | 75 % (62, 85) | 46 % (30, 63) |
| RC, % | 15 % | 10 % |
| RP, % | 59 % | 36 % |
| DoR par IRR | N = 44 | N = 18 |
| DoR médianne, mois (95 % IC) | 40.0 (23.1, NE) | 16.7 (7.4, NE) |
| % avec DoR ≥ 12 mois | 64 % | 44 % |
* Les
résultats d'une analyse de sensibilité considérant le nouveau traitement
anticancéreux comme un événement en plus de la progression et du décès sont de
23,1 mois chez les patients naïfs de traitement (14,8 ; NE) et de 12,0 mois
(6,3 ; NE) chez les patients précédemment traités.
N = nombre de patients ; TRO = Taux de réponse objective ; IC = Intervalle de Confiance ; RC = Réponse complète ; RP = Réponse partielle DoR = Durée de la Réponse ; IRR = Revue Radiologique Indépendante ; NE = non-évaluable
N = nombre de patients ; TRO = Taux de réponse objective ; IC = Intervalle de Confiance ; RC = Réponse complète ; RP = Réponse partielle DoR = Durée de la Réponse ; IRR = Revue Radiologique Indépendante ; NE = non-évaluable
Électrocardiographie
Dans l'analyse de tolérance des études poolées, l'incidence
de l'apparition d'un allongement de l'intervalle QTc
> 500 ms était de 1,1 % (4/363) dans l'ISP Combo450 (n = 372) et de 2,5 %
(5/203) dans le groupe de patients atteints de mélanome traité en monothérapie
par de l' encorafenib). Un allongement de
l'intervalle QTc > 60 ms comparé aux valeurs avant
traitement a été observé chez 6,0 % des patients (22/364) dans l'ISP Combo450
et chez 3,4 % (7/204) dans le groupe encorafenib
utilisé seul (voir la rubrique Propriétés pharmacodynamiques du RCP d'encorafenib).
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec le binimetinib dans un ou plusieurs sous-groupes de la population pédiatrique dans le mélanome (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
L'Agence européenne des médicaments a accordé la dispense de soumission des résultats des études réalisées avec le binimetinib dans tous les sous-groupes de la population pédiatrique dans le cancer du poumon (voir rubrique Posologie et mode d'administration - Population pédiatrique).
La pharmacocinétique du binimetinib a été étudiée chez des sujets sains et chez des patients présentant des tumeurs solides. Après une dose répétée de deux fois par jour concomitante avec l'encorafenib, l'état d'équilibre pour le binimetinib a été atteint dans un délai de 15 jours, sans accumulation majeure. La concentration maximale moyenne (CV %) était de 654 ng/ml (34,7 %) et l'exposition (aire sous la courbe) moyenne était de 2,35 µg.h/ml (28,0 %) en association à l'encorafenib, comme estimé par la modélisation de PK de population chez les patients atteints de mélanomes non résécables ou métastatiques porteur d'une mutation BRAF V600. La pharmacocinétique du binimetinib s'est révélée approximativement linéaire avec la dose.
Absorption
Après administration orale, le binimetinib est rapidement absorbé avec un Tmax médian de 1,5 heures. Après une dose orale unique de 45 mg [14C] de binimetinib chez des sujets sains, au moins 50 % de la dose de binimetinib ont été absorbés. L'administration d'une dose unique de 45 mg de binimetinib avec un repas riche en matières grasses et en calories a fait baisser la concentration maximale (Cmax) de 17 %, alors que l'aire sous la courbe (ASC) n'a pas changé. Une étude d'interaction médicamenteuse réalisée chez des sujets sains a indiqué que l'ampleur de l'exposition au binimetinib n'était pas altérée en présence d'un agent modifiant le pH gastrique (rabéprazole).
Distribution
Le binimetinib est lié aux protéines plasmatiques humaines à 97,2 % in vitro. La distribution du binimetinib est plus importante dans le plasma que dans le sang. Chez l'homme, le rapport sang/plasma est de 0,718. Après une dose orale unique de 45 mg [14C] de binimetinib chez des sujets sains, le volume apparent de distribution (Vz/F) du binimetinib est de 374 l.
Biotransformation
Après une dose orale unique de 45 mg [14C] de binimetinib chez des sujets sains, les principales voies de biotransformation du binimetinib observées chez l'homme comprennent la glycurono-conjugaison, la N-désalkylation, l'hydrolyse de l'amide et la perte d'éthane-diol de la chaîne latérale. La contribution maximale de la glycurono-conjugaison directe pour la clairance du binimetinib a été estimée à 61,2 %. Après une dose orale unique de 45 mg [14C] de binimetinib chez des sujets sains, environ 60 % de l'ASC de radioactivité circulante dans le plasma était attribuable au binimetinib. In vitro, CYP1A2 et CYP2C19 catalysent la formation du métabolite actif, ce qui cliniquement représente moins de 20 % de l'exposition au binimetinib.
Élimination
Après une dose orale unique de 45 mg [14C]
de binimetinib chez des sujets sains, une moyenne de 62,3 % de la
radioactivité a été éliminée dans les selles alors que 31,4 % était
éliminés dans l'urine.
Dans
l'urine, 6,5 % de la radioactivité a été excrétée sous forme de
binimetinib. La clairance apparente (CL/F) moyenne (CV en %) de
binimetinib était de 28,2 l/h (17,5 %). La demi-vie terminale médiane du binimetinib (T1/2) était de 8,66 h (8,10 à 13,6 h).
Interactions médicamenteuses
Effet des inducteurs ou des inhibiteurs de l'UGT1A1 sur le binimetinib
Le
binimetinib est principalement métabolisé via une glycurono-conjugaison
médiée par l'UGT1A1. Cependant, dans une sous-analyse de l'étude
clinique, il n'y avait aucune relation apparente observée entre
l'exposition au binimetinib et le statut de mutation de l'UGT1A1. De
plus, des simulations visant à étudier l'effet de 400 mg d'atazanavir
(inhibiteur de l'UGT1A1) sur l'exposition de 45 mg de binimetinib ont prédit une Cmax de binimetinib semblable en présence ou non d'atazanavir. L'étendue des interactions médicamenteuses médiées par l'UGT1A1 est donc minime, et probablement non cliniquement
pertinente ; toutefois, comme cela n'a pas été évalué dans une étude
clinique formelle, les inducteurs et les inhibiteurs de l'UGT1A1
doivent être administrés avec prudence.
Effet des enzymes CYP sur binimetinib
In vitro, CYP1A2 et CYP2C19 catalysent la formation du métabolite actif, AR00426032 (M3) par N-déméthylation oxydative.
Effet du binimetinib sur les substrats de CYP
Le binimetinib est un inhibiteur faible et réversible de CYP1A2 et de CYP2C9.
Effet des transporteurs sur binimetinib
Des expériences in vitro indiquent
que le binimetinib est un substrat de la p-glycoprotéine (P-gp) et de
la protéine de résistance au cancer du sein (BCRP, breast cancer
resistance protein). Il est peu probable que la P-gp ou la BCRP
engendrent une augmentation des concentrations de binimetinib
importante d'un point de vue clinique puisque le binimetinib présente
une perméabilité passive modérée à élevée.
Effet du binimetinib sur les transporteurs
Le
binimetinib est un inhibiteur faible de l'OAT3. Aucune interaction
médicamenteuse significative sur le plan clinique causée par le
binimetinib sur les autres transporteurs n'est attendue.
Le binimetinib est métabolisé par les UGT et CYP1A2, et est un substrat de la P-gp. Les inducteurs spécifiques de ces enzymes n'ont pas été étudiés et peuvent entraîner une perte d'efficacité.
Populations spéciales
Âge, poids corporel
Selon
une analyse pharmacocinétique de la population, l'âge ou le poids
corporel n'ont pas d'effet significatif sur le plan clinique sur
l'exposition systémique du binimetinib.
Sexe
D'après
une analyse pharmacocinétique (PK) de la population, le PK du
binimetinib était semblable chez les hommes et chez les femmes.
Race
Les
données ne sont pas suffisantes pour évaluer les différences
éventuelles au niveau de l'exposition au binimetinib selon la race ou
l'origine ethnique.
Insuffisance hépatique
Comme
le binimetinib est principalement métabolisé et éliminé par le foie,
les patients présentant une insuffisance hépatique modérée à sévère
peuvent être davantage exposés. Les résultats provenant d'une étude
clinique spécifique sur le binimetinib seul indiquent des expositions
similaires chez les patients présentant une insuffisance hépatique
légère (classe A de Child-Pugh) et chez les sujets dont la fonction
hépatique est normale. L'exposition totale au binimetinib (ASC) a été
doublée chez des patients présentant une insuffisance hépatique modérée
(classe B de Child-Pugh) à sévère (classe C de Child-Pugh) (voir
rubrique Posologie et mode d'administration). Cette
augmentation passe à trois fois plus en cas d'insuffisance hépatique
modérée à sévère lorsque l'on considère l'exposition en binimetinib
libre (voir rubrique Posologie et mode d'administration).
Maladie de Gilbert
Le
binimetinib n'a pas été évalué chez les patients atteints de la maladie
de Gilbert. La principale voie de transformation hépatique du
binimetinib étant la glycurono-conjugaison, la décision de traiter doit
être prise par le médecin traitant en tenant compte du rapport
bénéfice-risque individuel.
Insuffisance rénale
L'élimination
rénale du binimetinib est minime. Les résultats d'une étude clinique
spécifique ont montré que les patients souffrant d'une insuffisance
rénale sévère (DFGe ≤ 29 ml/min/1,73 m2), avaient eu une augmentation de 29 % de l'exposition (ASCinf), une augmentation de 21 % de la Cmax, et
une baisse de 22 % de la CL/F par rapport aux sujets sains
correspondants. Ces différences se situaient dans la variabilité
observée pour ces paramètres dans les deux cohortes de cette étude (25
% à 49 %) et la variabilité observée antérieurement dans des études
cliniques, c'est pourquoi ces différences sont peu susceptibles d'être
pertinentes sur le plan clinique.
Les effets de l'insuffisance rénale sur la pharmacocinétique du binimetinib en association à l'encorafenib n'ont pas été évalués sur le plan clinique.
Le binimetinib a un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Des troubles de la vision ont été rapportés chez certains patients traités par le binimetinib pendant les études cliniques. Il faut recommander aux patients de ne pas conduire de véhicules ou d'utiliser de machines en cas de troubles de la vision ou de tout autre effet indésirable qui pourrait nuire à leur aptitude à conduire des véhicules ou à utiliser des machines (voir rubriques Mises en garde spéciales et précautions d'emploi et Effets indésirables).
L'administration orale répétée de binimetinib chez le rat pendant une durée allant jusqu'à 6 mois était associée à une minéralisation des tissus mous, à des lésions de la muqueuse gastrique et à des changements minimes à modérés et réversibles de certains paramètres biochimiques à une dose correspondant à 7 à 12,5 fois l'exposition à la dose thérapeutique chez l'Homme. Dans une étude d'irritation gastrique chez le rat, une augmentation de l'incidence des lésions de la muqueuse superficielle et des ulcères hémorragiques a été observée. Chez les singes cynomolgus, l'administration orale de binimetinib était associée à une intolérance gastro-intestinale, à des changements modérés des paramètres biochimiques, à une hypercellularité de la moelle osseuse et à des lésions microscopiquesd'inflammation gastro-intestinale, réversibles aux plus faibles doses, inférieures aux expositions thérapeutiques humaines.
Le potentiel cancérogène du binimetinib n'a pas été évalué. Les études standard de génotoxicité avec binimetinib étaient négatives.
Les
effets potentiels du binimetinib sur le développement embryo-fœtal ont
été évalués chez le rat et le lapin. Chez le rat, un ralentissement du
gain de poids de la femelle gestante et une diminution du poids des
fœtus ont été notées un nombre réduit de sternèbres ossifiées a été
observé chez les fœtus. Aucun effet n'a été observé à 14 fois
l'exposition thérapeutique humaine.
Chez le lapin, de la mortalité, des signes cliniques de toxicité, un
plus faible poids corporel et des avortements ont été observés chez les
femelles gestantes. Le nombre de fœtus viables et le poids des fœtus
étaient réduits et les pertes et résorptions post-implantation
augmentées. Une augmentation de l'incidence des portées avec des fœtus
présentant des communications interventriculaires cardiaques et des
altérations du tronc pulmonaire a été notée à des élevées. Aucun effet
n'a été noté à une exposition correspondant à 3 fois l'exposition
thérapeutique humaine.
Aucune étude évaluant la fertilité n'a été menée avec le binimetinib. Dans les études de toxicité à doses répétées chez l'animal, aucun effet sur la fertilité n'a été mis en évidence à partir des examens histologiques des organes reproducteurs chez le rat et le singe.
Le binimetinib a un potentiel phototoxique in vitro.
Un risque minime de photosensibilisation a été mis en évidence in vivo à
une dose orale correspondant à une exposition 3,8 fois celle obtenue à
la dose thérapeutique humaine. Ces données indiquent que le binimetinib
présente un risque minime de réaction phototoxique aux doses
thérapeutiques pour les patients.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation locale en vigueur.
Liste I.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription hospitalière.
Prescription réservée aux médicins compétents en CANCEROLOGIE.
Prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription hospitalière.
Prescription réservée aux médicins compétents en CANCEROLOGIE.
Prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE.
Remboursement en fonction de l'indication (JO du 22/08/2019) :
La seule indication thérapeutique ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie est le traitement en association à l'encorafenib de patients adultes atteints de mélanome non résécable ou métastatique porteur d'une mutation BRAF V600.
Comprimé pelliculé (comprimé).
Comprimés
pelliculés jaune à jaune foncé, non sécables, biconvexes, de forme
ovale d'environ 12 mm de longueur et 5 mm de largeur, gravé avec le
logo « A » sur un côté du comprimé et « 15 » de l'autre côté.
Plaquette en PVC/PVDC/Alu contenant 12 comprimés. Chaque boîte contient 84 comprimés.
Chaque comprimé pelliculé contient 15 mg de binimetinib.
Excipient à effet notoire
Chaque comprimé pelliculé contient 133,5 mg de monohydrate de lactose.
Pour la liste complète des excipients, voir rubrique Liste des excipients
Comprimé
Monohydrate de lactose
Cellulose microcristalline (E460i)
Silice colloïdale anhydre (E551)
Croscarmellose sodique (E468)
Stéarate de magnésium (E470b)
Pelliculage
Alcool (polyvinylique) (E1203)
Macrogol 3350 (E1521)
Dioxyde de titane (E171)
Talc (E533b)
Oxyde de fer jaune (E172)
Oxyde de fer noir (E172)